The [4π + 2π] Cycloaddition (Diels-Alder Reaction)
- Thermal concerted [4π + 2π] cycloaddition
- Main features of the [4π + 2π] cycloaddition
- Stereoselectivity: Preference for endo transition structure
- Matching the reacting components: Effects of donor, acceptor and conjugating substituents
- Regioselectivity
- Lewis acid catalysis
- Steric effects in the diene
1 Thermal concerted [4π + 2π] cycloaddition
The archetypal Diels-Alder reaction is a [4π + 2π] cycloaddition in which a diene (the 4π component) combines with a dienophile (the 2π component) to produce a 6-membered ring. The process is very useful because it generates four new sp3 carbons stereoselectively, the geometry of both starting molecules being conserved within the product (the reaction is stereospecific). For example, if the dienophile bears two groups with a cis relationship, the two groups will remain cis in the product. The net reaction involves the loss of two pi bonds and the gain of two sigma bonds, which makes it about 167 kJ/mol (40 kcal/mol) exothermic. The six-membered transition structure for the process resembles a folded cyclohexane (boat conformation). Figure 2.1 depicts the cycloaddition using both line structures and orbitals; note that the symmetry of the interacting orbitals permits synchronous bond formation while the p orbitals of the pi bonds become the sigma bonds of the product.

Nobel Prize 1950

Nobel Prize 1950
![FMO interactions in the [4π + 2π] cycloaddition of 1,3-butadiene to ethene](images_PR/2003.png)
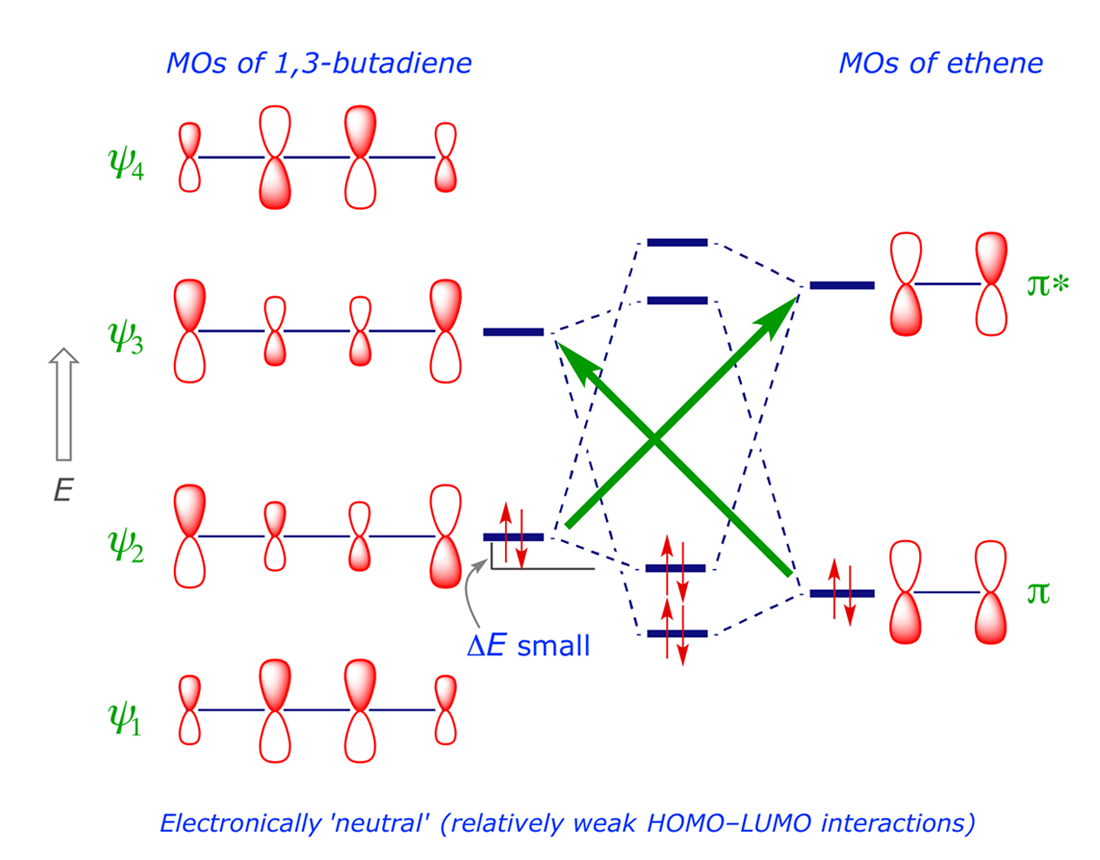
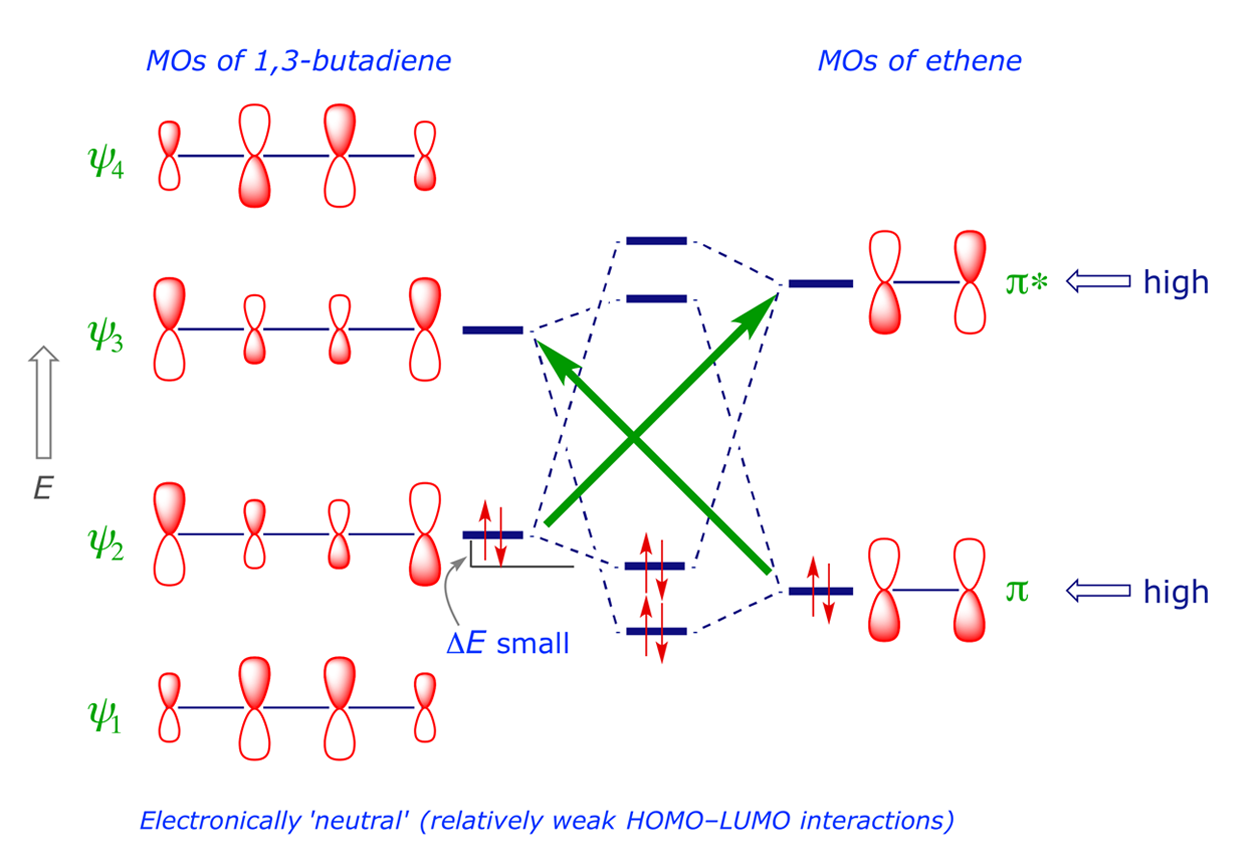
The orbital interaction shown in Figure 2.1 is between the HOMO(4π) and the LUMO(2π). The symmetry properties of these orbitals are correctly 'matched' for the simultaneous overlap of each bonding pair, i.e. the interacting lobes are in phase. The same is true of the LUMO(4π) and HOMO(2π) combination (Figure 2.2). For ground-state molecules, the frontier orbitals are thus correctly matched for synchronous bond formation, making this a thermally allowed [4π + 2π] cycloaddition.
Overall reaction (X = Y = H):
![Simplified mechanistic scheme for a [4π + 2π] cycloaddition](images_PR/2012.png)
FMO interactions:
![HOMO-LUMO interactions in a [4π + 2π] cycloaddition](images_PR/2015.png)
The complete orbital interaction diagram is constructed by linear combination of the starting systems, butadiene and ethene (Figure 2.3). The green arrows indicate the two HOMO-LUMO interactions; note that ΔE is small because the HOMO-LUMO energy gap is relatively large (see the relevant equation here).
2 Main features of the [4π + 2π] cycloaddition
• Concerted (synchronous) mechanism, compatible with orbital symmetry requirements for bonding overlap, no polar intermediates, reversible. ΔG for the cycloreversion process becomes more favorable (negative) with increased temperature because the −TΔS term dominates over the ΔH term (ΔG = ΔH − TΔS).
![Diels-Alder [4π + 2π] dimerisation of cyclopentadiene](images_PR/2021.png)
• Stereoselective, endo product is favoured over exo product (kinetic control).
![[4π + 2π] Cycloaddition of cyclopentadiene to methyl acrylate](images_PR/2024.png)
• Matching of components, the 'normal' reaction involves electron-poor 2π and electron-rich 4π components.
![[4π + 2π] Cycloaddition of butadiene to maleic anhydride](images_PR/2027.png)
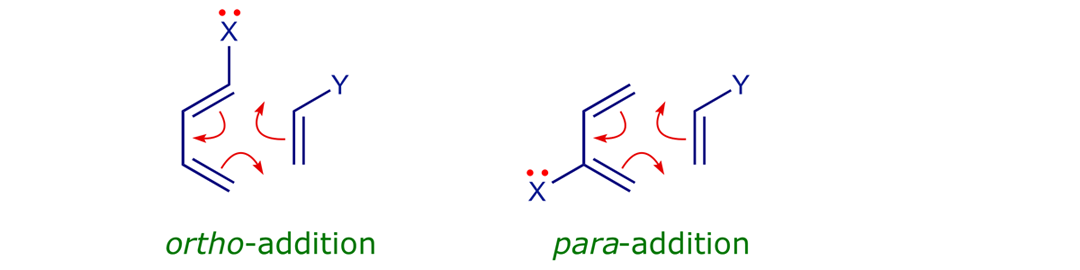
• Regioselective, typically as shown below (X = donor, Y = acceptor).
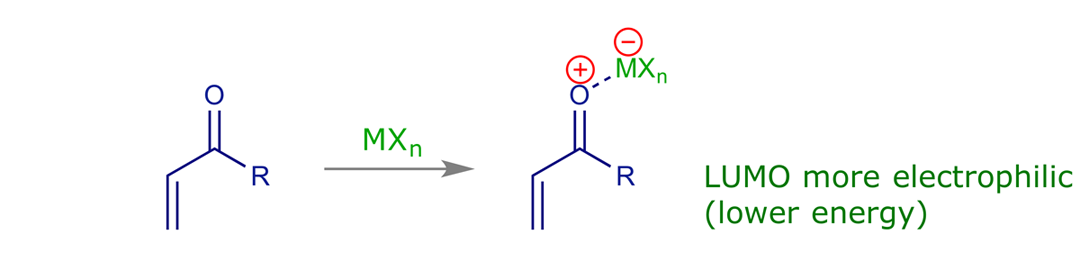
• Lewis acid catalysis can make the reaction faster and more selective (coordinates to 2π component, lowers its LUMO energy).
3 Stereoselectivity: Preference for endo transition structure
The Diels-Alder cycloaddition tends to give much higher yields if an electron-withdrawing group is conjugated with the dienophile π bond. The major product is the one with the electron-withdrawing group underneath, endo, rather than exo, in which it is less sterically hindered. Favorable interactions between the electron-withdrawing group and the diene (*), known as secondary orbital interactions, have been invoked to rationalise this endo selectivity. In one of the classic illustrations of this phenomenon, cyclopentadiene reacts with methyl acrylate (010) with a diastereoselectivity of 9:1 in favour of the endo product (Figure 2.4). The generalised stereochemical outcome of the endo- and exo-cycloaddition modes in substituted reaction partners is shown in Figure 2.5.
![[4π + 2π] Cycloaddition of cyclopentadiene to methyl acrylate](images_PR/2036.png)
![Generalised scheme showing endo- and exo-selection in [4 + 2] cycloadditions](images_PR/2037.png)
The Diels-Alder dimerisation of two butadiene molecules (011) is another example of endo selectivity. The endo transition structure has an additional interaction between the unfilled diene ψ3 MO and the filled diene ψ2 MO (Figure 2.6). Note once again that the symmetry properties of the interacting frontier orbitals allow both the Diels-Alder reaction itself and the secondary orbital overlap which favours the endo transition structure.
![Secondary orbital overlap in the [4π + 2π] dimerisation of 1,3-butadiene](images_PR/2039.png)
4 Matching the reacting components: Effects of donor, acceptor and conjugating substituents
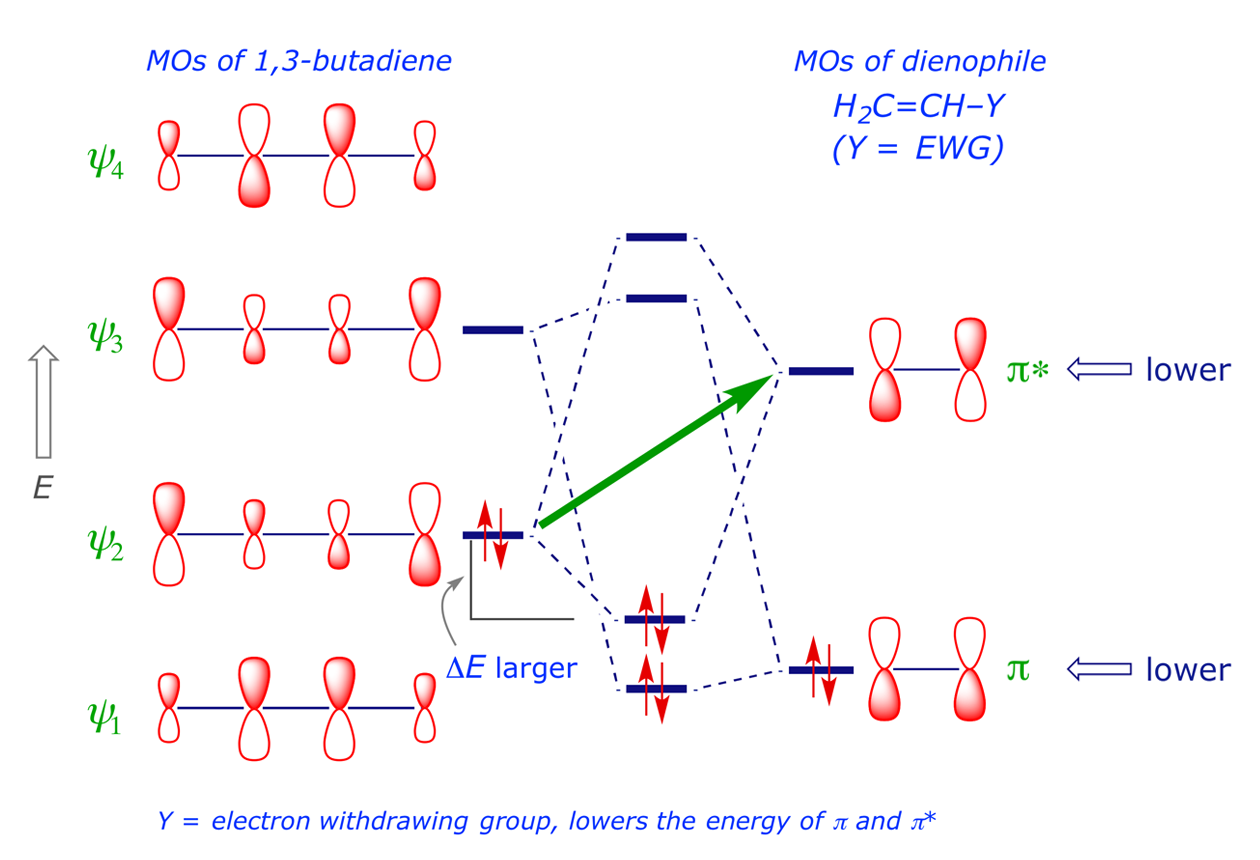
The soft-soft interaction of filled with empty orbitals is the major attractive force in the Diels-Alder transition structure, which is stabilised through two HOMO-LUMO interactions: The filled ψ2 of the diene interacts with the unfilled ψ* of the dienophile, while the empty ψ3 of the diene interacts with the filled π of the dienophile. For 1,3-butadiene reacting with ethene (Figure 2.3), the HOMO-LUMO interactions are relatively weak since the interacting orbitals are not close in energy (ΔE is small). The interaction increases if the ψ2–ψ* gap is made smaller, usually by using a dienophile with an electron attractor group Y. Substituents on the diene or dienophile change the energy of the FMOs and polarise their attached π bond, introducing a rate-increasing hard-hard (Coulombic) factor into the reaction and enhancing the regioselectivity (Figure 2.7).
To see the effect of donor and acceptor groups on the orbital energy of a π system, we can compare the limiting cases (Y = CH2− as donor; Y = CH2+ as acceptor). These are represented by the HOMO and LUMO energy levels in ethene, allyl cation and allyl anion. The right-hand side of Figure 2.8 shows what happens if a good π donor is attached to a double bond. The π donor HOMO interacts with both the HOMO and the LUMO of the double bond to produce an allylic system with a new HOMO and LUMO, both of which are raised in energy relative to those of the double bond (Figure 2.8, green upward arrows). In the general case, the HOMO energy of a π system is raised by attached donor substituents.
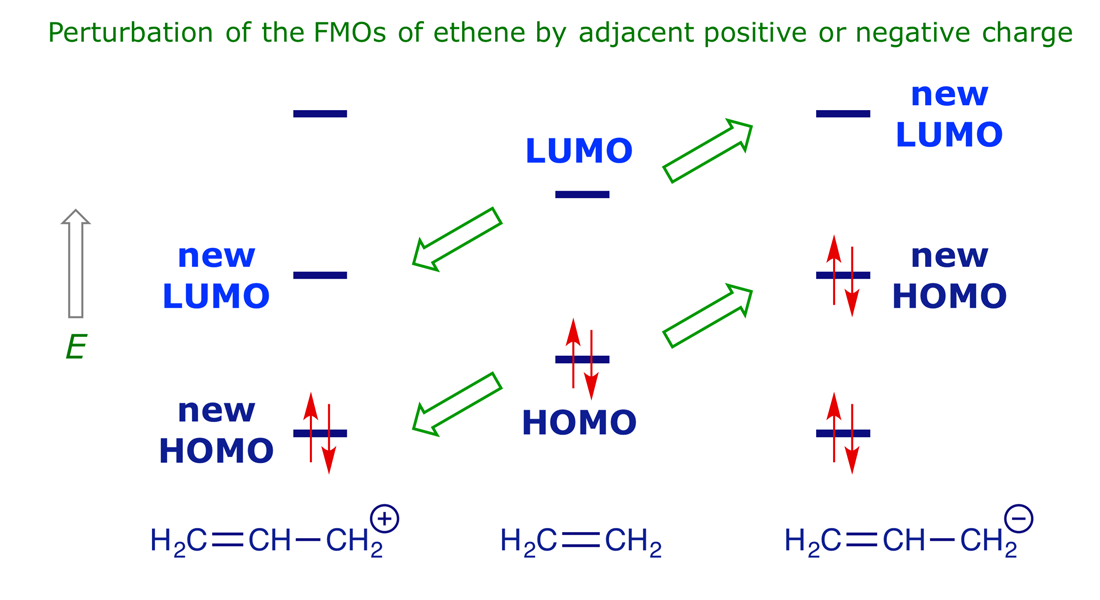
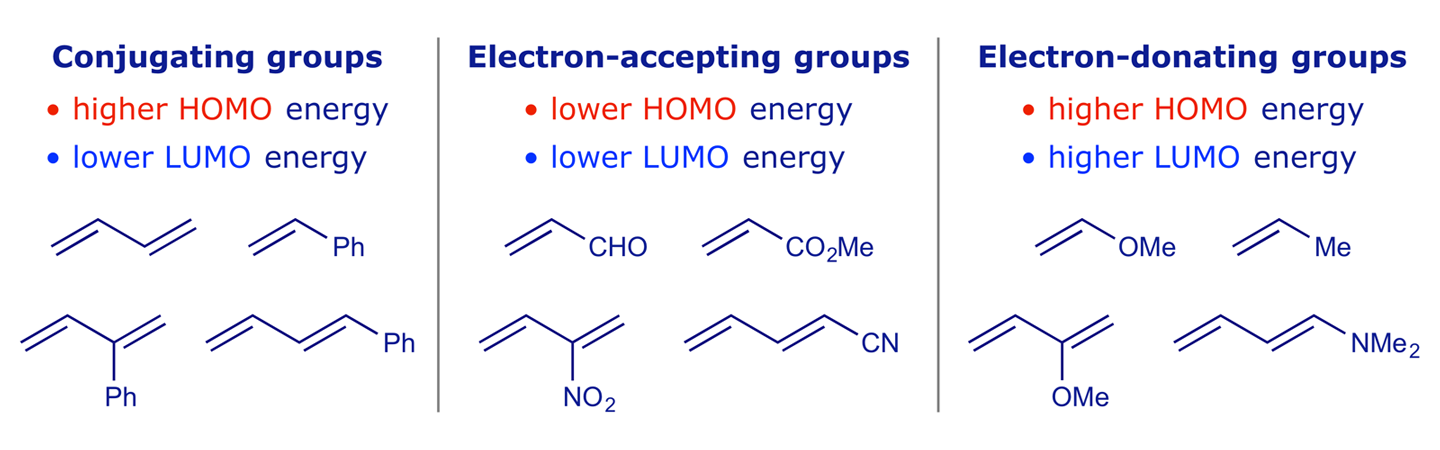
The left-hand side of Figure 2.8 shows what happens if a good π acceptor is attached to a double bond. The LUMO of the π acceptor interacts with both the HOMO and the LUMO of the double bond to produce an allylic system with a new HOMO and LUMO, both of which are lowered in energy relative to those of the double bond (Figure 2.8, green downward arrows). In the general case, the LUMO energy of a π system is lowered by attached acceptor substituents. When the LUMO energy of a π system is lower, then it is softer, and the stabilisation resulting from overlap with the HOMO of another π system is greater. The effects of substituents on the frontier orbital energies of various alkenes and dienes are summarised below in Figure 2.9.
5 Regioselectivity
The 'normal' Diels-Alder matching pair is an electron-rich diene with an electron-deficient dienophile, so the dominant FMO interaction is HOMO(4π)–LUMO(2π). The π orbitals of butadiene and ethene are readily polarisable and influenced by donor or acceptor substituents. When X is a donor and Y is an acceptor, the π MOs polarise and the orbital coefficients change as shown below. A prediction of the regioselectivity of a cycloaddition is obtained by matching the largest coefficients in the diene HOMO and dienophile LUMO (the greater the overlap, the stronger the covalent bonding).
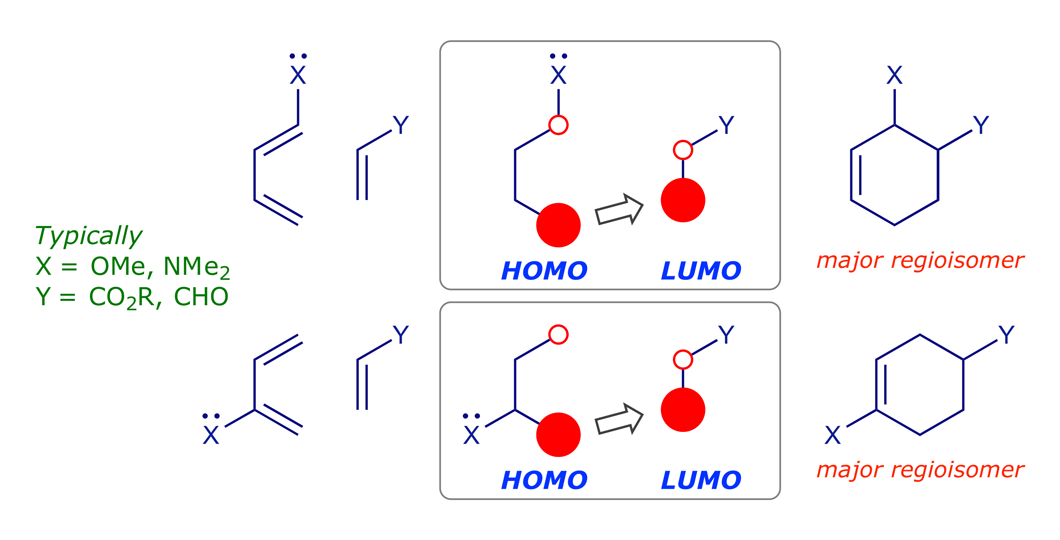
Good illustrations are provided by reactions 012, 013 and 014; note the different reaction conditions (temperature, time). Groups directly attached to C(1) and C(4) of the diene, i.e. the reaction termini, have a more pronounced effect than groups attached to C(2) or C(3).
![[4π + 2π] Cycloaddition of 1-(diethylamino)-1,3-butadiene to ethyl acrylate](images_PR/2057.png)
![[4π + 2π] Cycloaddition of 2-methoxy-1,3-butadiene to methyl acrylate](images_PR/2060.png)
![[4π + 2π] Cycloaddition of Danishefsky's diene to maleic anhydride](images_PR/2063.png)
6 Lewis acid catalysis
Dienophiles are more electrophilic and more polarised when coordinated to Lewis acids, which are electron withdrawing. The dienophile LUMO is more polarised and its energy is lower, increasing both the reaction rate and the regioselectivity of the reaction.
![Uncatalysed and catalysed [4π + 2π] cycloadditions of 2-methyl-1,3-butadiene to methyl acrylate](images_PR/2066.png)
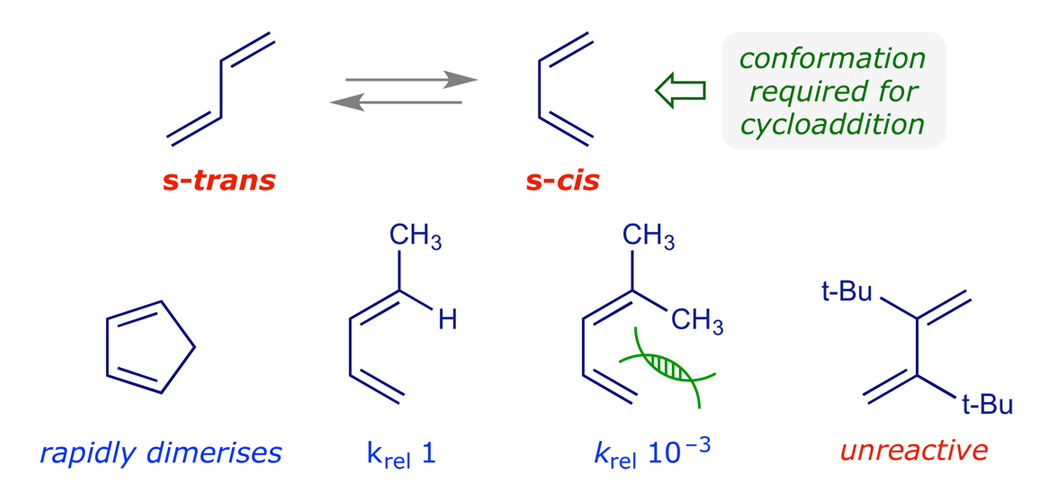
7 Steric effects in the diene
For an open-chain diene to undergo a Diels-Alder cycloaddition it must first adopt the s-cis conformation. As a cyclic diene, 1,3-cyclopentadiene is locked in this conformation and consequently very reactive. In contrast, some dienes are unreactive because they are either slow or unable to adopt the required conformation for steric reasons.