7 Analysis of enantiomers
7.1 Enantiomeric purity and the enantiomeric excess (e.e.) scale
A 50:50 mixture of two enantiomers is called a racemic mixture, and is optically inactive. When there is only one enantiomer present, a substance is described as optically pure or homochiral. Between racemic and optically pure lies a range of unequal mixtures of two enantiomers, with one enantiomer present in excess. Although in this situation there will be a measurable rotation in a polarimeter, it will not be the full value of the specific rotation [α]D but an intermediate value depending on the composition of the mixture. Optical activity is linear, so the situation can be shown graphically as a straight-line, as below for lactic acid.
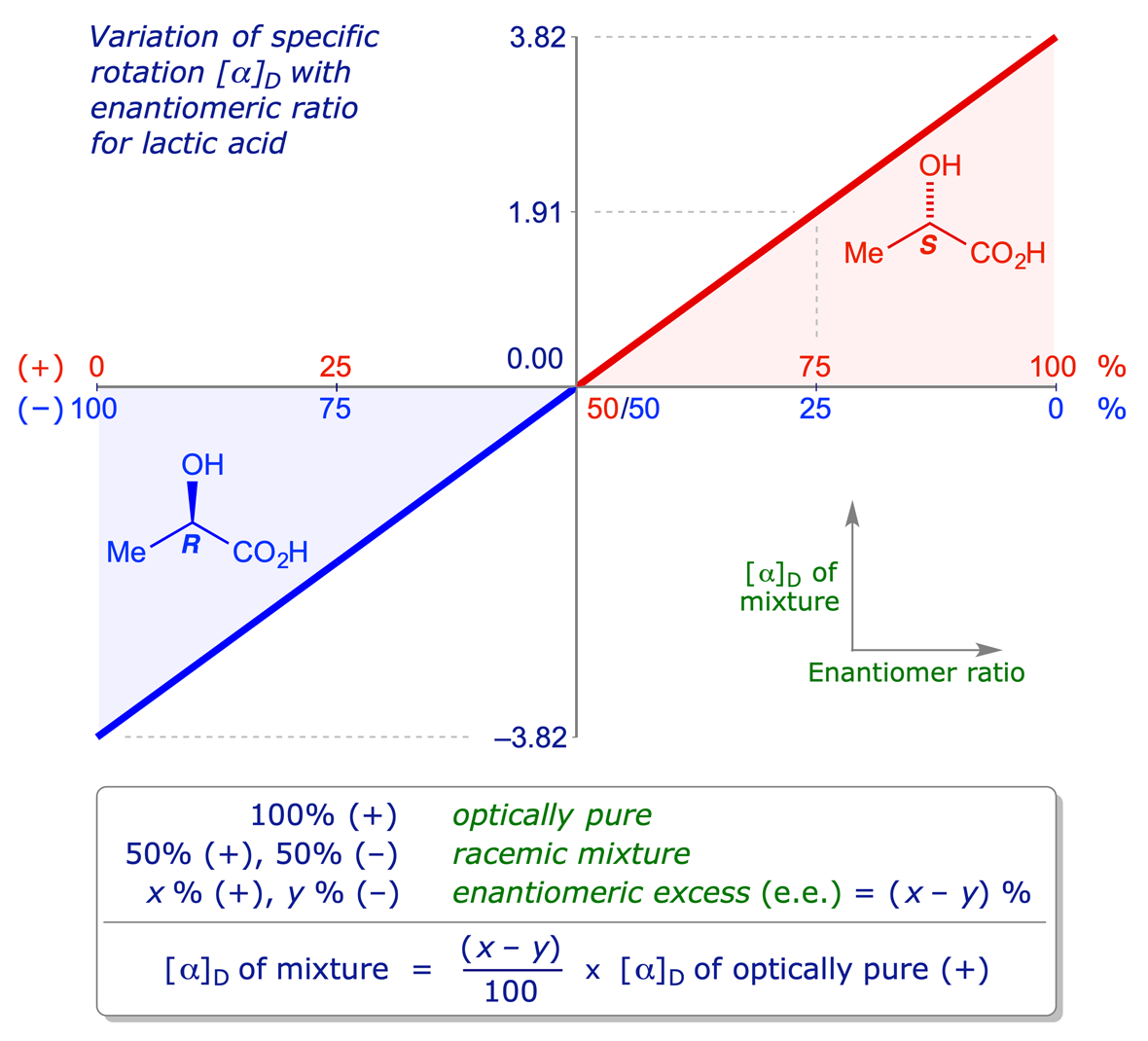
For example, a mixture consisting of 75% (+)-lactic acid and 25% (−)-lactic acid is said to contain a 50% enantiomeric excess of the (+)-enantiomer.
To calculate the proportions of each enantiomer from an e.e. value, remember that the difference between the % optical purity and 100% is made up of racemic substance, half of which is one enantiomer and half the other enantiomer.
Example:
An e.e. value of 60% means 60% optically pure + 40% racemic mixture. The '40% racemic' fraction consists of 20% of each enantiomer, so the ratio of the two enantiomers is major (60+20)%, minor 20%, i.e. 80:20.
7.2 Determining the enantiomeric excess (e.e.) of a non-racemic compound
7.2.1 Measuring e.e. by polarimetry
In the previous section it was shown how the optical purity of non-racemic lactic acid could be calculated by measuring the specific rotation of the sample and comparing the value obtained with that of optically pure lactic acid. However, in practice there are a number of problems associated with the accuracy of polarimetry measurements which limit their usefulness as an indicator of optical purity.
- It may not be possible to measure a rotation value accurately if the [α]D value is small (it can even be zero) or only a small quantity of sample is available.
- The [α]D of the pure enantiomer may never have been accurately established.
- The sample must be completely free of impurities, both chiral and achiral.
- For some compounds there is a non-linear relationship between e.e. and [α]D.
For the above reasons optical rotation measurements should, at best, only be used to obtain a rough estimate of the e.e. value of a non-racemic mixture of enantiomers. In general, greater accuracy can be obtained by one of the methods described below.
7.2.2 Measuring e.e. via sample derivatisation
The principle of derivatisation for assay purposes is the same as that for the preparative resolution of enantiomers (section 6.2). The enantiomeric mixture in question is reacted with an enantiomerically pure derivatising agent to convert the enantiomeric relationship into a diastereoisomeric one. The mixture can then be analysed by any technique which can be used to analyse diastereoisomers quantitatively (e.g. NMR spectroscopy, HPLC, GLC).
Typical derivatisations:
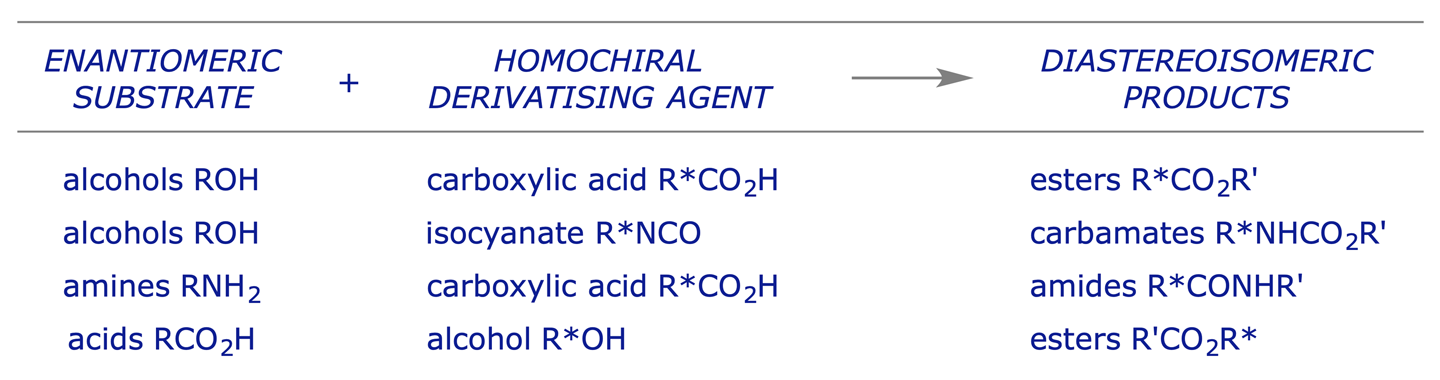
Some acids used for forming diastereoisomeric derivatives:
An example involving the use of Mosher's reagent is shown below. The Mosher acid chloride (R)-1 is prepared from Mosher's acid, and then used to 'derivatise' a mixture of enantiomeric amines 2. This gives a mixture of amides 3 and 4 which are diastereoisomers and so can be analysed by HPLC or NMR spectrocopy. An advantage of Mosher's system is the presence of the CF3 group, which allows both 1H and 19F NMR spectroscopy to be used for the assay.
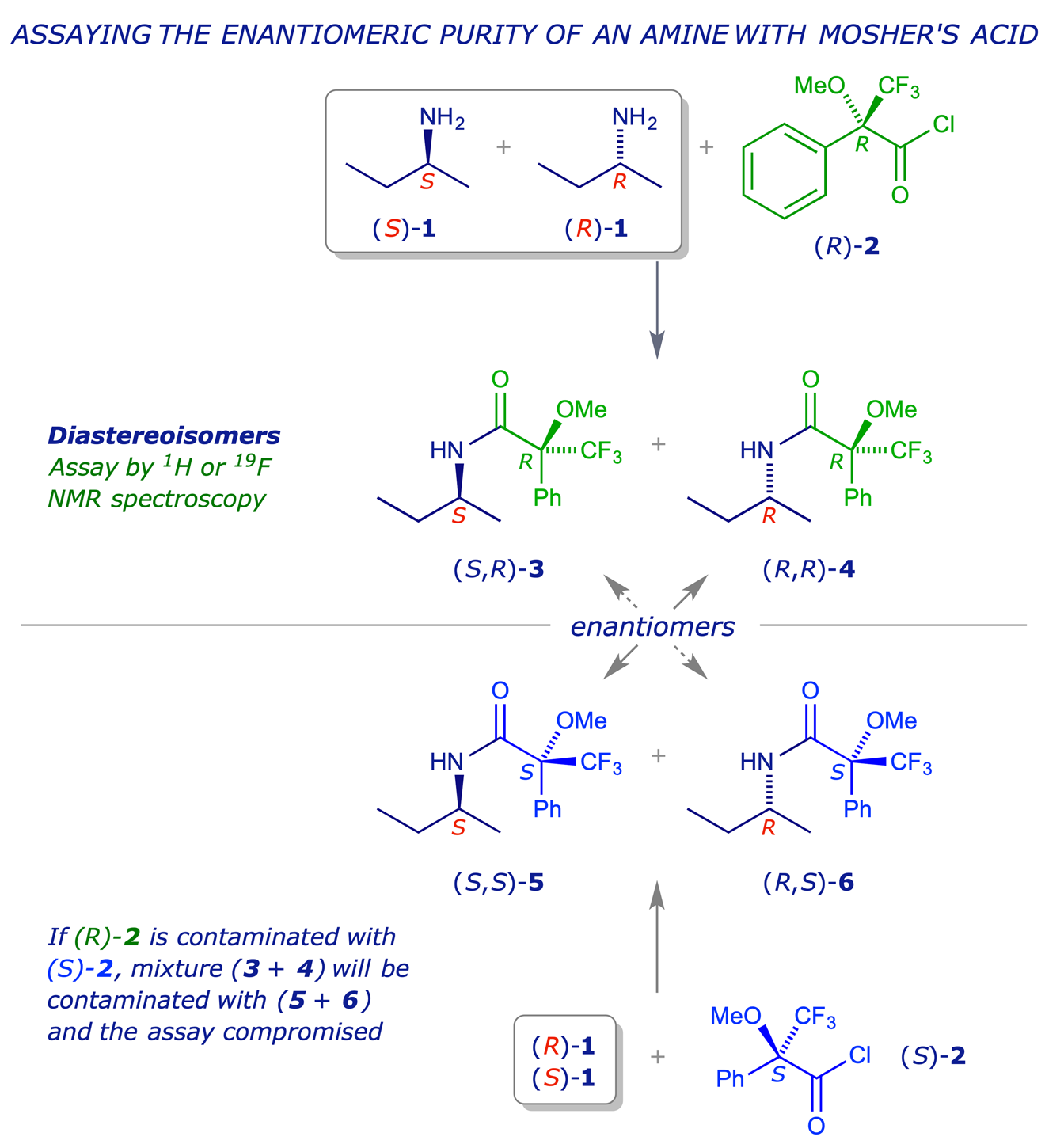
It is important to ensure that the derivatising agent, in this case (R)-1, is enantiomerically pure, because the two amides 5 and 6 which would be formed from the enantiomer (S)-1 would be enantiomers of the amides 4 and 3 respectively, and would therefore be indistinguishable from them by HPLC or NMR, leading to a false result. Other requirements of this strategy are as follows:
- All of the starting material must have reacted with the derivatising agent before the analysis, in order to be certain that the diastereoisomer ratio exactly matches that of the enantiomers in the original mixture. This is because the chiral derivatising agent may react faster with one enantiomer than the other (kinetic resolution).
- The crude derivatised product must be analysed without purification, to avoid the possibility that the ratio of the diastereoisomers is changed during purification (remember that diastereoisomers are separable by chemical purification techniques such as chromatography or crystallisation).
- Test the derivatisation and analysis on a racemic mixture of enantiomers before attempting to analyse any optically active material. This will verify that the procedure works properly if it indicates a 50:50 ratio of enantiomers.
- Use accurate analytical techniques (e.g. high field NMR spectroscopy, HPLC or GLC) for measuring the ratios of the derivatised (diastereoisomeric) products.
7.2.3 Chiral chromatography of enantiomeric mixtures
Nowadays enantiomers are routinely analysed by passing them through chromatography columns containing chiral materials. Chromatographic separation relies on a difference in affinity between a stationary phase, usually silica, and a mobile phase – the eluting solvent in high performance liquid chromatography (HPLC) or the carrier gas in gas-liquid chromatography (GLC) – mediated by weak interactions (H-bonding, dipole-dipole or van der Waals). A stationary phase can be made chiral simply by bonding it with an enantiomerically pure compound.
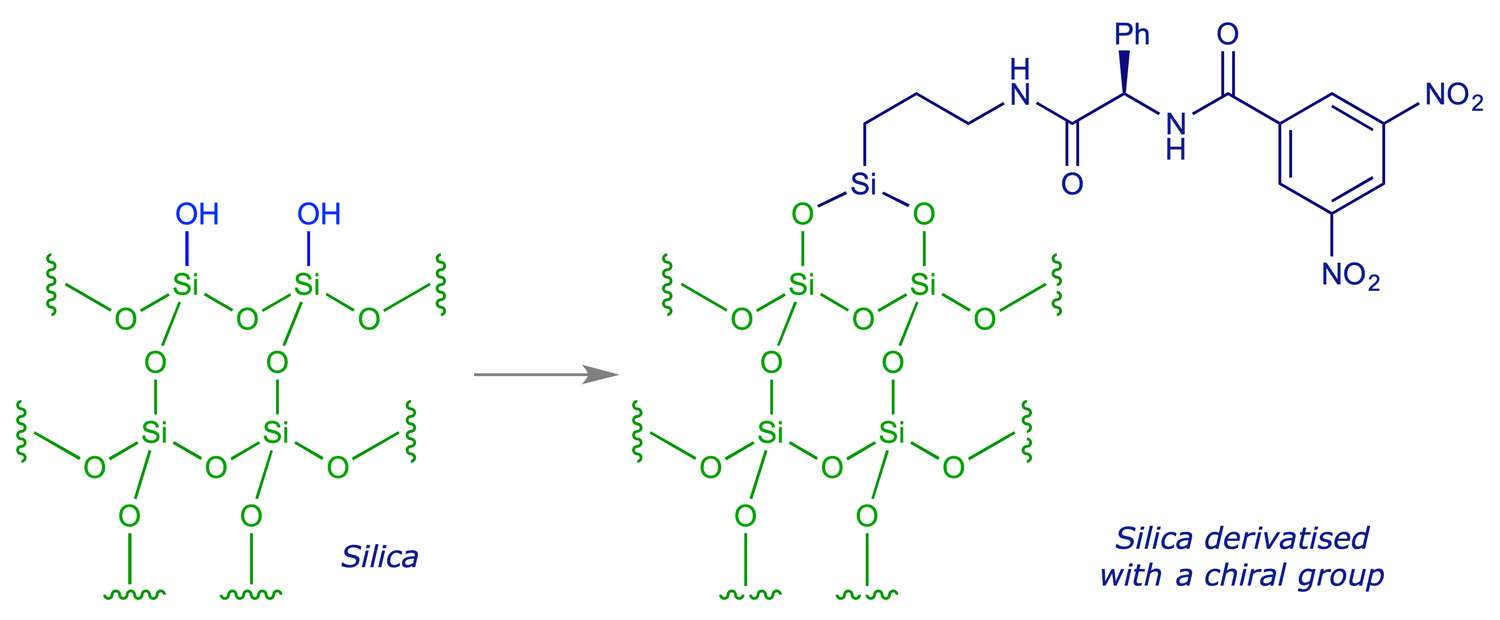
As two enantiomers interact with a homochiral stationary phase, the interactions of each enantiomer with the stationary phase have a diastereoisomeric relationship. If the difference in the interactions is sufficient, there will be a separation of the enantiomers (the enantiomer which binds more strongly to the stationary phase will take longer to pass through the column). As the enantiomers emerge (at different times) from the column they enter a UV detector (high sensitivity; recall that the enantiomers have the same UV spectrum), and the signals are integrated to provide theassay.
Chiral gas liquid chromatography (GLC) works on the same principle, and is one of the best methods for analysing enantiomer mixtures. The equipment is expensive, but the method is rapid and sensitive (detection limits of better than 0.01% of a minor enantiomer are not uncommon).
7.2.4 Chiral additives used in high-field NMR spectroscopy
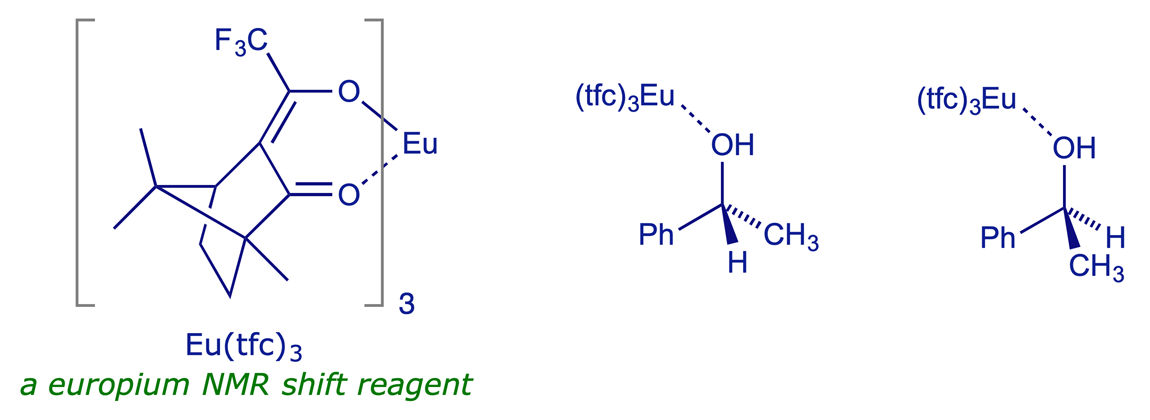
Operationally one of the simplest methods for assaying a pair of enantiomers is to add a chiral shift reagent or a chiral solvating agent to the mixture when recording its NMR spectrum. Chiral shift reagents are lanthanide complexes, and as mild Lewis acids – lone pair acceptors – they readily coordinate to heteroatoms e.g. O (or N) in enantiomeric alcohols (or amines). As they are chiral, there is a diastereoisomeric relationship between the complexing agent and each enantiomer of the compound being analysed, and in principle the two resulting diastereoisomeric complexes will have different NMR spectra. As long as comparable signals from the two diasteroisomeric complexes can be completely distinguished (ideally with 'baseline separation'), they can be accurately integrated and the ratio of enantiomers thereby estimated.
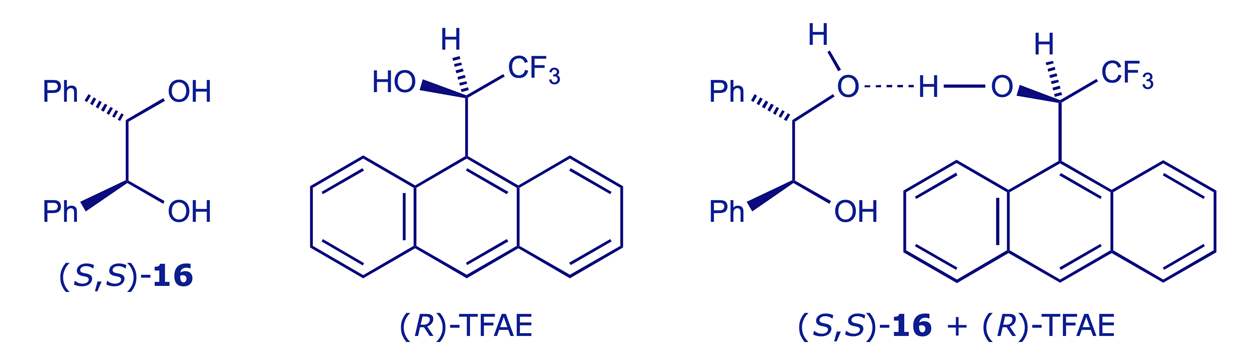
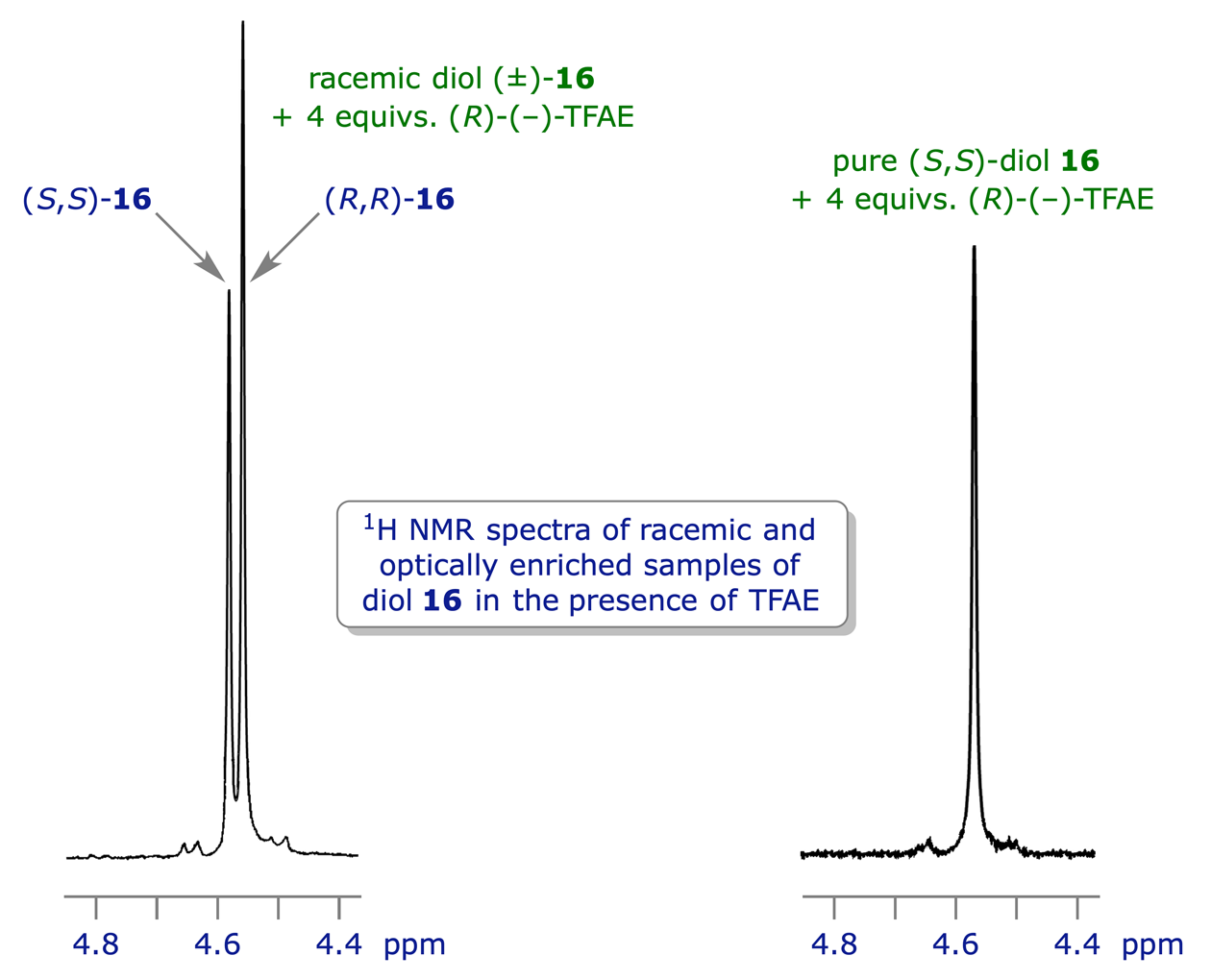
The most widely used chiral solvating agent is 2,2,2-trifluoro-1-(9-anthryl)ethanol (TFAE), which can also coordinate (via dipole-dipole or H-bonding interactions) to lone pairs e.g. those on O or N. Combining a single enantiomer of TFAE with the two enantiomers of a chiral compound will give two coordination complexes, and these will be diastereoisomers. If the spectra from the two complexes are distinguishable they can be assayed to determine the original ratio of enantiomers. In the example shown below, (−)-TFAE is used to establish the enantiomeric purity of a sample of enantiomerically enriched 1,2-diphenylethane-1,2-diol (hydrobenzoin) 16.